AAV Therapy of Novel Serotype cc47 Achieves Biochemical Correction of the Liver in Murine Glycogen Storage Disease Type Ia
Name
Anna McKane
Major
Molecular Bioscience / Cell and Molecular Biology
Class
2023
About
Anna McKane is from the U.S. and majored in Cell and Molecular Biology. She is interested in translational and veterinary medicine.
Signature Work Project Overview
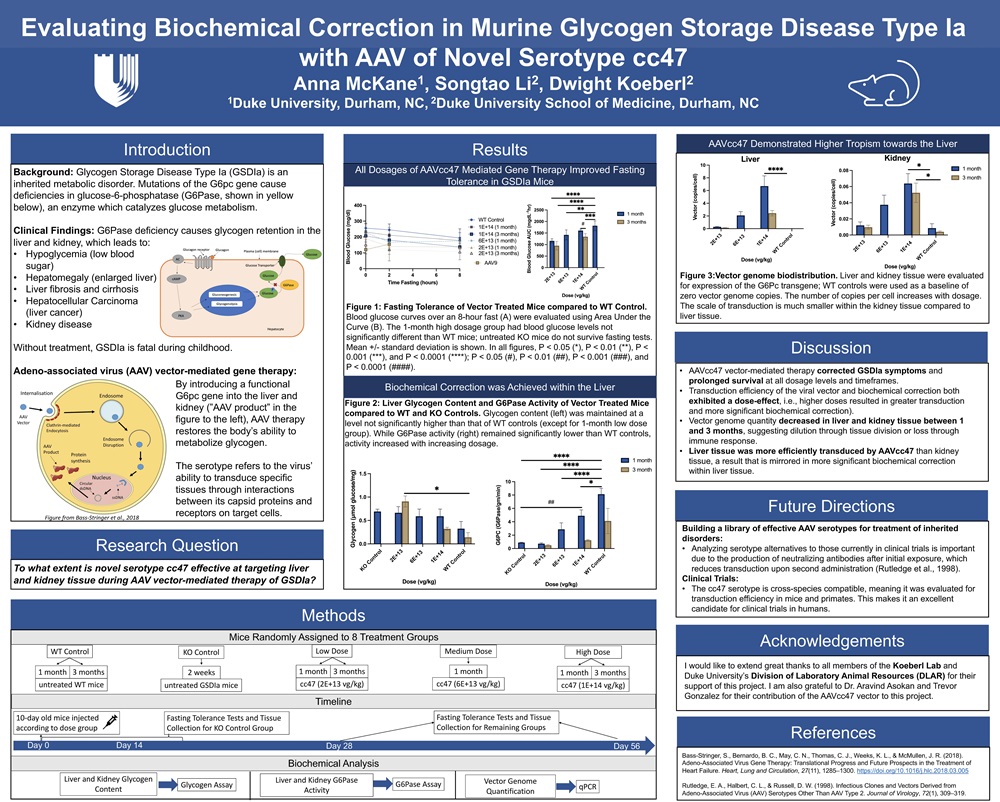
Glycogen Storage Disorder Type Ia (GSDIa) is an inherited metabolic disorder characterized by deficiency in glucose-6-phosphatase (G6PC), an enzyme critical to gluconeogenesis and glycogenolysis. Unable to metabolize glycogen, GSDIa-affected individuals are at increased risk of chronic hypoglycemia as well as renal and hepatic conditions. The scope and severity of symptoms has motivated the development of gene replacement therapy. My Signature Work project contributes to this investigation by evaluating efficacy of gene therapy mediated by the novel AAV serotype cc47 in treating murine GSDIa. This serotype – which refers to the virus’ ability to transduce specific tissues through interactions between its capsid proteins and receptors on target cells- has been engineered to be cross-species compatible, making it particularly appealing for future clinical trials. AAV.cc47 vector was administered to G6Pase (-/-) mice in five treatment groups of varying duration and dosage. The therapy prolonged survival and increased fasting tolerance at all dosage levels. Quantification of vector genome biodistribution suggested the cc47 serotype demonstrates higher tropism towards the liver than the kidney, which was mirrored by greater hepatic biochemical correction. Liver G6PC activity appeared to be higher than in untreated mice in both the medium and high dosage groups, though not achieving statistical significance. Liver glycogen content was maintained at levels not significantly different than those found in WT tissue. By evaluating the serotype cc47 for the first time in vivo, this study develops a preliminary understanding of its tissue tropism and contributes to a bank of AAV serotypes that may be useful for future preclinical studies and clinical trials.